2019年以来的新型冠状病毒(2019-nCoV, SARS-CoV-2),目前在全球大流行,截至2022年4月24日全球累计5亿多确诊病例,已有620多万人因感染SARS-CoV-2死亡 (来源于https://covid19.who.int/),而逃避宿主免疫的新型冠状病毒突变株(VOC)的陆续出现,对全球经济和公共卫生带来空前的挑战。新冠疫情发生以来,国际上多个研究小组针对SARS-CoV-2进行了系列药物研发工作。目前在临床试验登记平台官方网站(clinicaltrials.gov)上有7871份临床试验记录。理想的抗病毒药物应该针对涉及SARS-CoV-2生命周期的必需蛋白质。 木瓜蛋白酶样蛋白酶(PLPro)和主蛋白酶(Mpro/3CLpro)是新型冠状病毒产生的两种重要的蛋白酶。SARS-CoV-2病毒进入细胞后,核衣壳与病毒自身的正义链RNA释放到细胞质中,正义链RNA的开放阅读框ORF1a和ORF1b翻译产生非结构蛋白,由PLpro和 Mpro/3CLpro参与裂解,最终产生包括螺旋酶(Helicase)和RNA依赖性RNA聚合酶(RdRp)在内的16种非结构蛋白,参与病毒转录、复制过程[1]。PLpro还介导抗病毒蛋白的去泛素化和去ISG化,从而参与调节宿主抗病毒先天免疫[2,3]。因此,抑制PLPro和Mpro/3CLpro能够有效抑制病毒的感染与复制,是抗病毒药物研发的关键靶点。
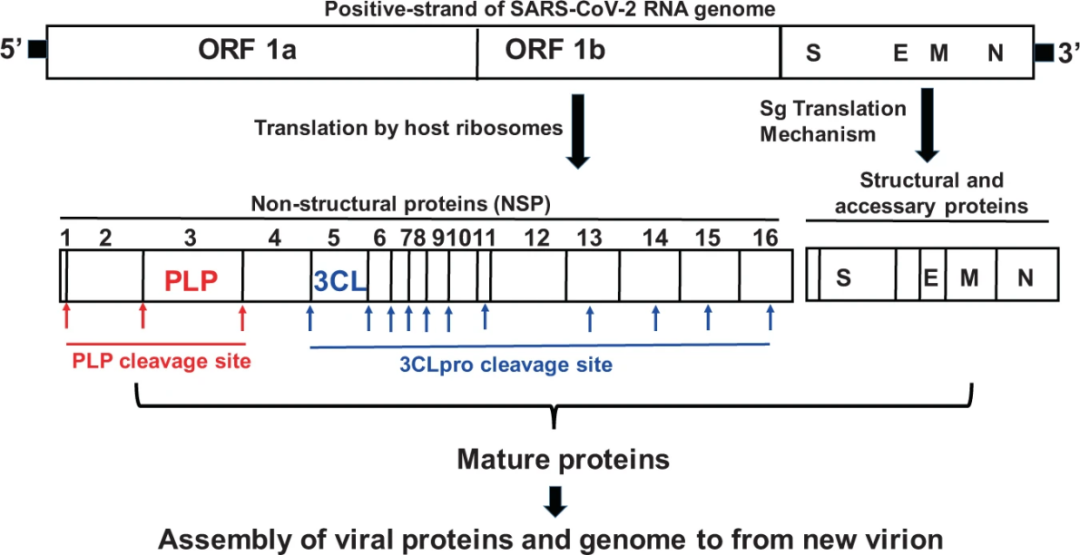
新冠病毒核心酶PLPro
PLpro位于nsp3,在三个位点切割病毒多聚蛋白pp1a和pp1ab,产生nsp1、nsp2和nsp3。如图2[4],PLpro包含一个N端泛素样结构域(Ubl)和一个催化核心结构域。催化核心结构域包括三个子结构域:拇指(Thumb)、手掌(Palm)和手指(Finger),像张开的右手一样。底物结合位点位于拇指结构域和手掌结构域之间的溶剂暴露裂缝中,具有由C111、H272和D286组成的催化三元体。底物结合位点识别保守序列LXGG↓X(底物的氨基酸残基编号为P4-P3-P2-P1↓-P1',向下的箭头表示裂解位点)。PLpro的亚位点S1-S4分别为底物的P1-P4提供了结合位点。在催化位点附近有一个6-氨基酸(残基267-272)的柔性环,称为封闭环2(blocking loop 2,BL2),在控制底物进入活性位点方面起着重要作用。
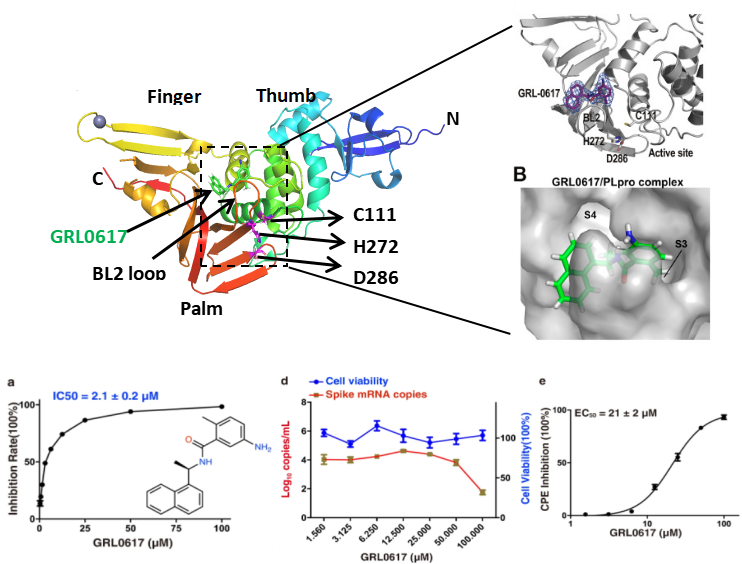
研究者筛选和评价了一批FDA批准的针对SARS和MERS等 PLpro的临床药物对SARS-CoV-2 PLpro的适用性,发现一种萘类的非共价抑制剂GRL0617显示出了很强的抑制活性,IC50约为2.0 μM,EC50约为20 μM,并且在Vero E6细胞的抗病毒实验中表现出了较好的抑制效果。GRL0617通过占据和封锁PLpro底物结合口袋S3和S4(图2),发挥了抑制活性,BL2环向抑制剂方向关闭,干扰肽基序LXGG的识别。而结合位点与PLpro的活性中心无关[4-9]。 随后,Osipiuk等报道了几个GRL0617类似物,它们对SARS-CoV-2 PLpro也表现出良好的抑制作用[7]。Ma等报道了两个优化后的Jun9-75-4和Jun9-72-2,IC50(分别为0.62和0.67μM)和EC50(分别为7.88和6.62μM)比GRL0617低几倍,从而使它们成为非常有效的PLpro抑制剂[10]。Shen等通过探索GRL0617之外的其他结合位点对GRL0617的效价进行了优化,发现2 -苯基噻吩(phenylthiophene)抑制剂XR8-23和 XR8-24对SARS-CoV-2 PLpro IC50值分别为0.39和0.56μM,在人类肺上皮A549细胞抗病毒感染的EC50值分别为1.4和1.2μM,比GRL0617有很大改进[11]。高通量筛选还发现了一种新的先导药物YM155,对SARS-CoV-2 PLpro的抑制IC50值为2.47 μM,EC50值为0.17 μM[12]。从组合底物库中鉴定到的拟肽抑制剂VIR250和VIR251是SARS-CoV-2 PLpro的共价抑制剂,这两种共价抑制剂对SARS-CoV和SARS-CoV-2 PLpros均表现出良好的抑制作用,对人去泛素酶几乎没有交叉反应[13]。GRL0617使BL2保持在一个关闭的构象中,而VIR251则是使BL2稳定在开放构象中,基于此又筛选了几种具有类似相互作用模式的潜在抑制剂[14,15]。 另外,最近有研究者筛选到了一种具有广谱抗冠状病毒活性的非共价先导抑制剂F0213,IC50 值为7.4 µmol/L,EC50 值为4.5 µmol/L,可有效的抑制SARS-CoV、SARS-CoV-2(包括Omicron)、MERS-CoV、hCoV-229E和hCoV-OC43等多种冠状病毒(图3)。既能通过阻断病毒多蛋白裂解来抑制冠状病毒复制,又能通过拮抗PLpro去泛素酶活性来促进抗病毒免疫,口服给药的F0213具有一定的临床应用潜力[16]。
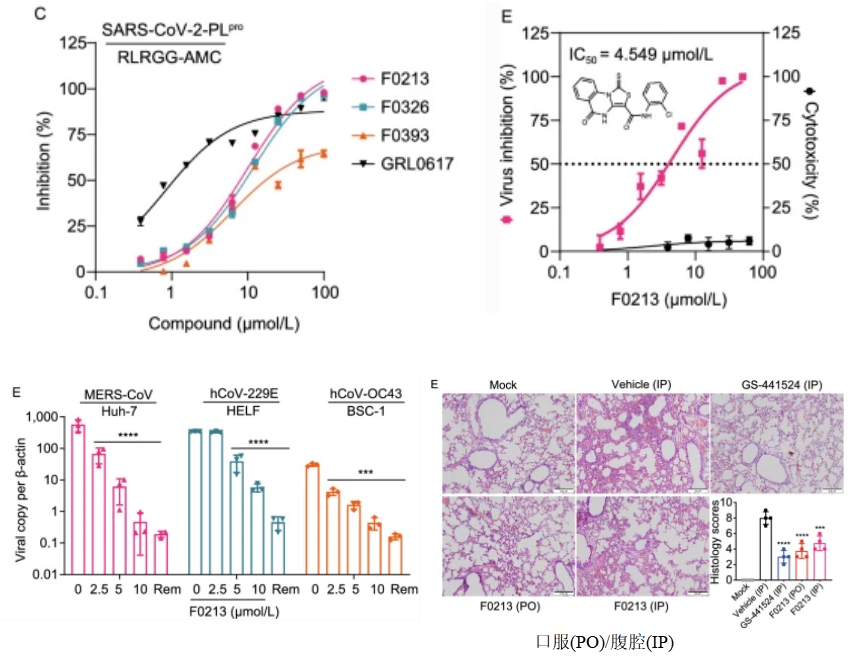
图3. F0213对 PLpro的抑制作用
目前,在clinicaltrials.gov可查到两种PLpro抑制剂已进入临床研究。一种是用于治疗严重痤疮的药物异维A酸,已在四项临床试验中测试其治疗新冠病毒的潜力(试验号为:NCT04361422、NCT04382950、NCT04389580和NCT04353180)。另一种是FDA批准的药物Ebselen,由于PLpro的半胱氨酸残基对其酶活性至关重要,Ebselen能与半胱氨酸残基共价结合,阻断冠状病毒PLpro的活性。(试验号为:NCT04483973和NCT04484025)
新冠病毒核心酶Mpro/3CLpro
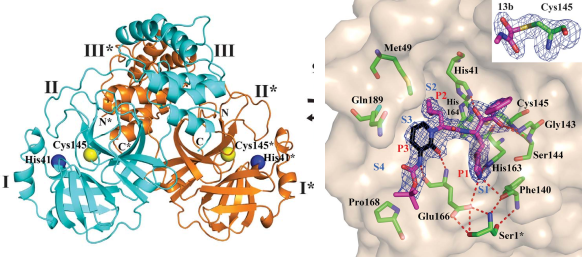
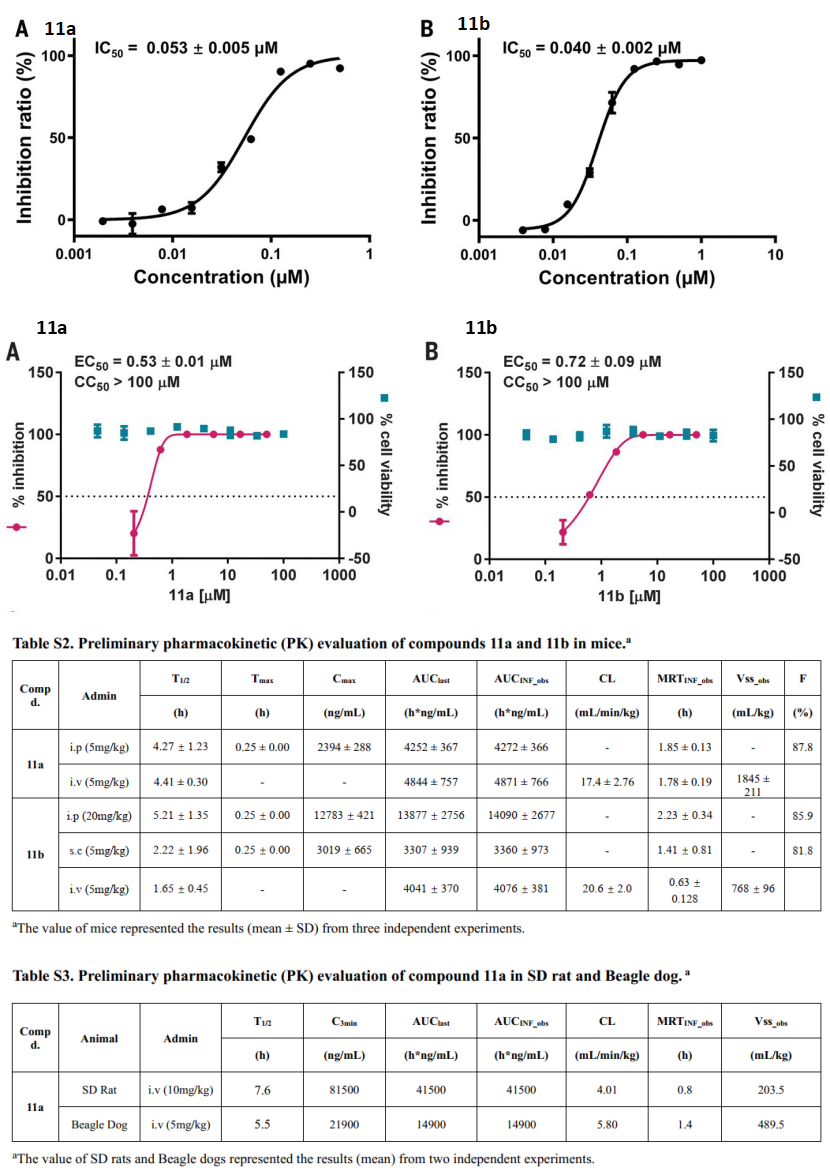
Mpro位于nsp5,在多聚蛋白1ab上至少有11个裂解位点。Mpro大多数底物的识别顺序为Leu-Gln↓(Ser, Ala, Gly) (↓为裂解位点)。底物的氨基酸残基编号为P4-P3-P2-P1↓-P1 ',Mpro的亚位点S1-S4分别为底物的P1-P4提供了结合位点。上海药物研究所解析了Mpro的结构(图4),Mpro的活性形式是同源二聚体,每个原体由三个域组成(结构域I、II和III)。结构域I(残基8-101)和II(残基102-184)由反平行的β-桶组成,结构域I和II一起形成类胰凝乳蛋白酶的结构。结构域III(201- 306)主要由5个α-螺旋组成,排列成一个大的球状簇,结构域III负责二聚化。抑制剂α-ketoamide对MERS-CoV、SARS-CoV和肠道病毒均具有抗病毒作用。两个化合物11a和11b是α-ketoamide 的衍生物,具有较好的抗病毒活性。另外,解析的化合物11a和11b与主蛋白酶的晶体结构表明,底物结合口袋位于结构域I和II之间的裂口。Mpro识别底物的p4-p1’位置[17]。
FRET的实验结果表明,11a和11b具有良好的抑制效果,IC50分别为0.053μM和0.040 μM。在Vero-E6细胞中,对SARS-CoV-2的EC50分别为530 nM和720 nM。且静脉注射时候,小鼠、大鼠和比格犬表现出良好的药代动力学性质(图5)[18]。11a是有望作为抗病毒药物往前推的,上海药物研究所官网给出的11a编号为DC402234,转让给的前沿公司的编号为FB2001,目前该化合物有2项临床试验在进行,试验号为:NCT04766931和NCT05197179。
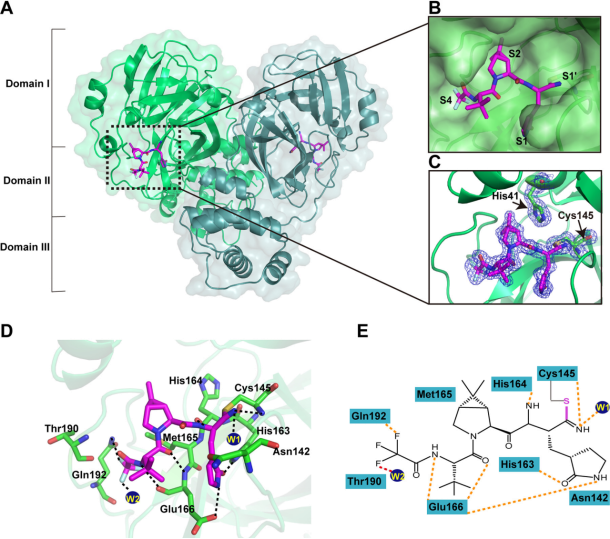
许多已经被发现的Mpro抑制剂表现出一系列的结合机制。这些抑制剂通常是模仿天然肽底物的模拟肽,弹头主要为Michael受体、醛类和不同类型的酮类等。它们与Mpro中的C145残基共价结合,发挥抑制作用,应该对冠状病毒具有普遍的广谱活性。临床批准的药物carmofur[19]和boceprevir[20]被重新利用,对SARS-CoV-2 Mpro表现出低的微摩尔抑制活性。临床前抑制剂GC376[20]显示出抑制活性。比较这些抑制剂-Mpro复合物结构,发现除了 carmofo和boceprevir外,这些小分子都与Mpro催化残基半胱氨酸形成C-S共价键,并在P1位置形成内酰胺环,与S1亚位点非常匹配。SARS-CoV-2 Mpro亚位点S2似乎更喜欢疏水相互作用。然而,这些抑制剂的P3位置与S3口袋匹配得不太好。将来可能会进行结构优化,以研究更适合该亚位点的基团。 2021年12月22日,辉瑞宣布美国FDA批准其新型COVID-19口服抗病毒候选药物Paxlovid 的紧急授权申请(EUA),用于治疗非住院、具有发展成重症疾病高风险成人COVID-19感染。根据EPIC-HR 研究,该药可以将住院或死亡风险降低89%。辉瑞Paxlovid主要成分之一是PF-07321332。PF-07321332与SARS-CoV-2 Mpro复合物结构(图6)可见,在SARS-CoV-2 Mpro结构域I和II之间裂隙中的狭长空腔具有多个抑制剂结合位点,其中PF-07321332占据亚位点S1、S2和S4,除了典型的共价相互作用外,PF-07321332还与活性位点形成多种非共价相互作用[21]。
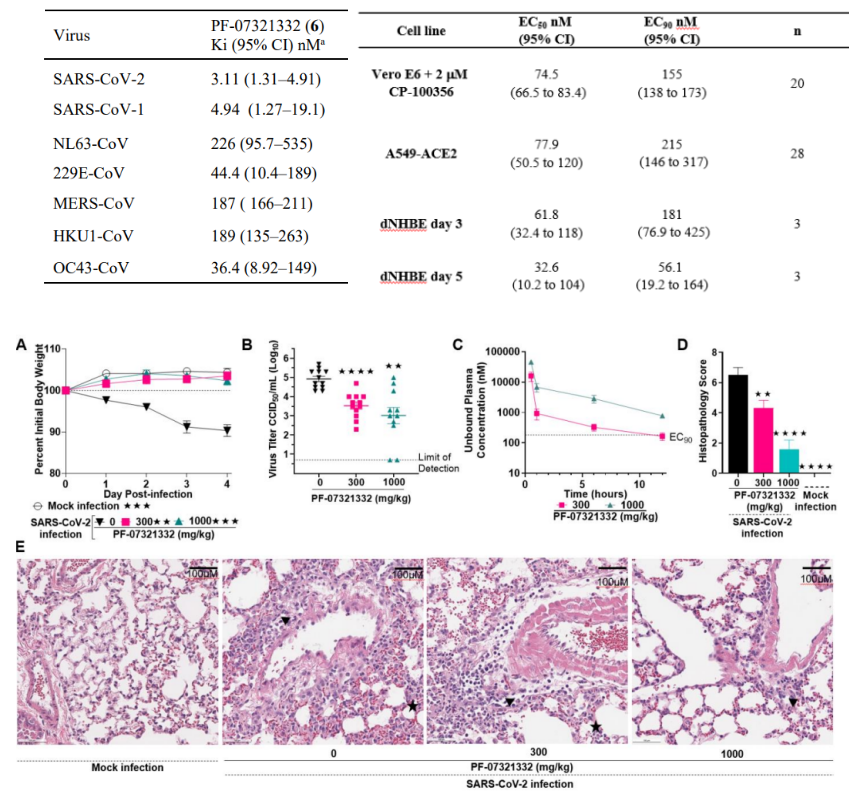
PF-07321332对所有已知感染人类的冠状病毒类型的FRET Mpro检测显示了有效的抑制作用,其中对SARS-CoV-2 Ki(95%CI)值为3.1nM(图7)。另外,用两种与病理相关的细胞模型:表达ACE2的人腺癌来源的肺泡基底上皮细胞(A549)和分化正常的人支气管上皮细胞(dNHBE)来评估PF-07321332的体外抗病毒活性,EC50分别为77.9nM和61.8nM,表明在两种细胞模型中PF-07321332对病毒复制都有强烈抑制效果(图7)。使用小鼠适应SARS-CoV-2的动物模型,评估PF-07321332的体内抗病毒活性,结果证明,在之前体外实验观察到的抗病毒浓度下,它在体内也可以有效降低感染鼠肺部病毒浓度,同时能够明显改善感染小鼠的多灶性肺部病变(图7)[22]。
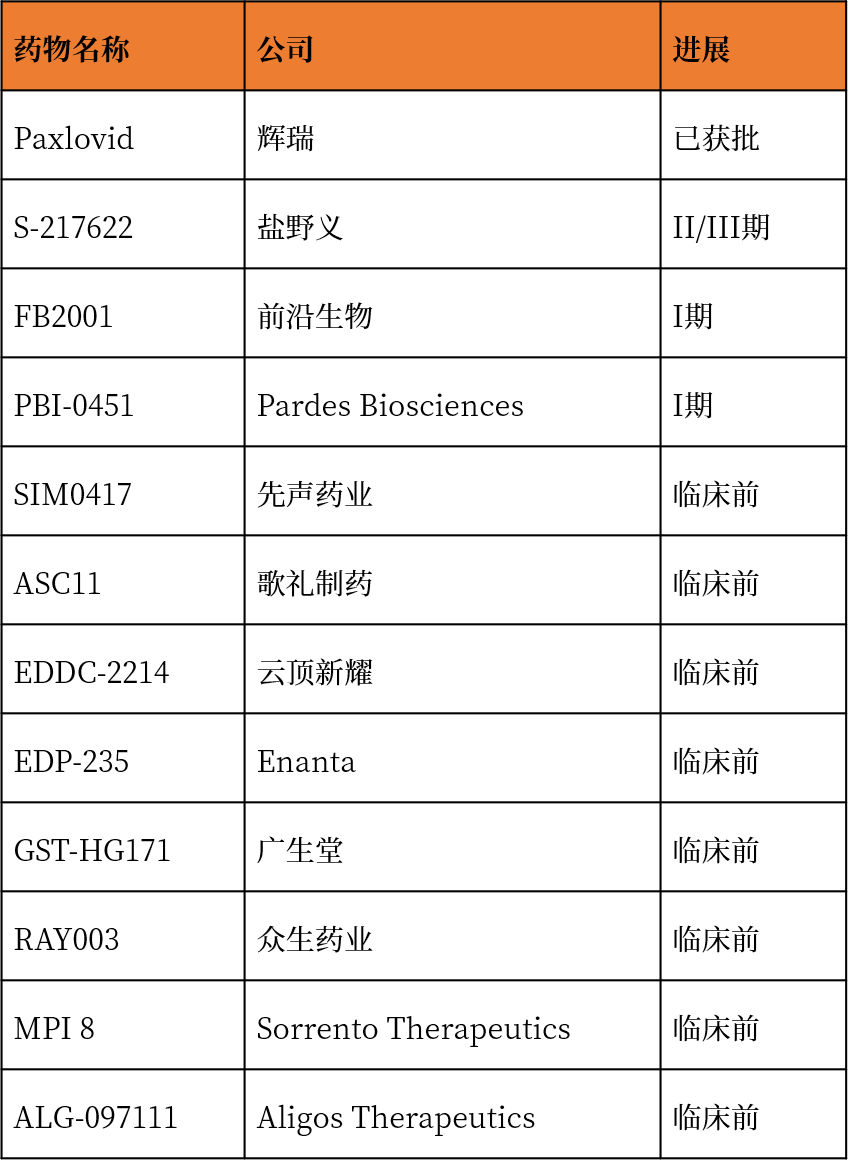
另外,药物再利用是药物发现的重要策略。一些现有的广谱抗病毒药物,如双抑制剂,双硫仑(disulfiram)/依布硒(ebselen),能同时抑制Mpro和PLpro的水解作用,目前正在进行临床试验。随着SARS-CoV-2的高传染性在全球范围内不断爆发,药物再利用、新导识别和基于结构的新导优化的综合应用有望进一步加快研发进程,开发安全的特异性抑制剂。总之,尽管已有一些令人鼓舞的结果,但SARS-CoV-2药物的发现仍远不能满足需求。未来的研究仍需努力,最终开发出有前景的临床药物。
扫描下面二维码即可申请PLpro/3CLpro酶活检测方法,只需要15min,大大节省实验时间,为PLpro/3CLpro以及该酶抑制剂的评估助力。

产品推荐
Recombinant SARS-CoV-2 3C-like Proteinase/3CLpro (Cat.No.:CR92)
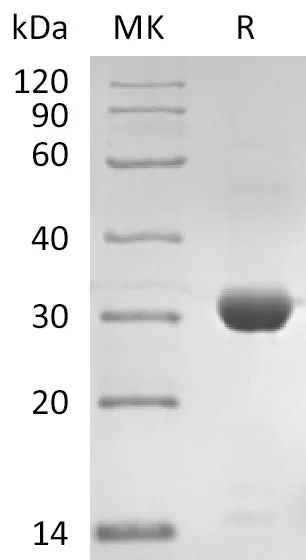
Purity:>95%(SDS-PAGE)
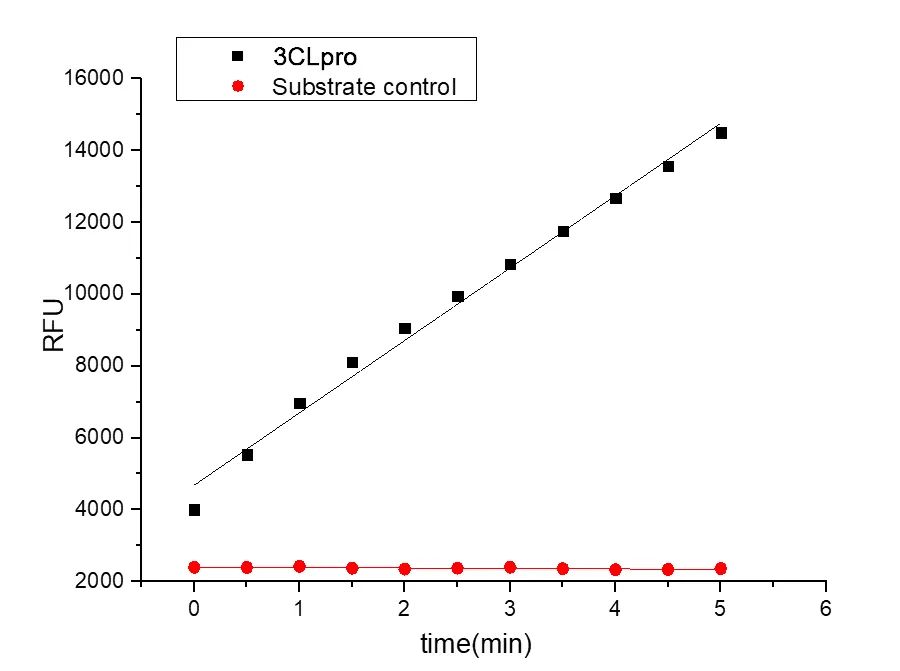
Bioactivity: Measured by its ability to cleave a fluorogenic peptide substrate, Dabcyl-KTSAVLQSGFRKME-Edans (Synthetic).The specific activity is 136.6pmol/min/μg.
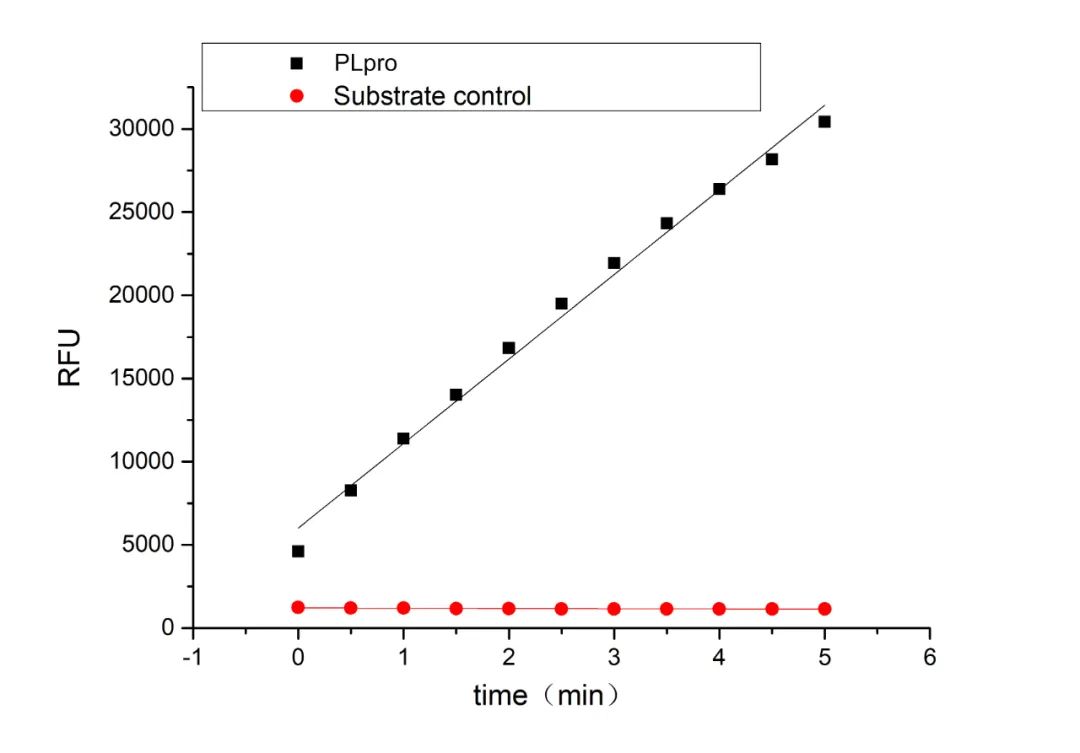
Recombinant 2019-nCoV Papain-Like Protease/PLPro (Cat.No.:CR77)
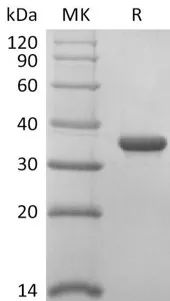
Purity:>95%(SDS-PAGE)
[1] Mody, Vicky et al. “Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents.” Communications biology vol. 4,1 93. 20 Jan. 2021, doi:10.1038/s42003-020-01577-x
[2] Békés, Miklós et al. “Recognition of Lys48-Linked Di-ubiquitin and Deubiquitinating Activities of the SARS Coronavirus Papain-like Protease.” Molecular cell vol. 62,4 (2016): 572-85. doi:10.1016/j.molcel.2016.04.016
[3] Shin, Donghyuk et al. “Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity.” Nature vol. 587,7835 (2020): 657-662. doi:10.1038/s41586-020-2601-5
[4] Gao, Xiaopan et al. “Crystal structure of SARS-CoV-2 papain-like protease.” Acta pharmaceutica Sinica. B vol. 11,1 (2021): 237-245. doi:10.1016/j.apsb.2020.08.014
[5] Osipiuk, Jerzy et al. “Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors.” Nature communications vol. 12,1 743. 2 Feb. 2021, doi:10.1038/s41467-021-21060-3
[6] Fu, Ziyang et al. “The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery.” Nature communications vol. 12,1 488. 20 Jan. 2021, doi:10.1038/s41467-020-20718-8
[7] Osipiuk, Jerzy et al. “Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors.” Nature communications vol. 12,1 743. 2 Feb. 2021, doi:10.1038/s41467-021-21060-3
[8] Klemm, Theresa et al. “Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2.” The EMBO journal vol. 39,18 (2020): e106275. doi:10.15252/embj.2020106275
[9] Freitas, Brendan T et al. “Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease.” ACS infectious diseases vol. 6,8 (2020): 2099-2109. doi:10.1021/acsinfecdis.0c00168
[10] Ma, Chunlong et al. “Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay.” ACS central science vol. 7,7 (2021): 1245-1260. doi:10.1021/acscentsci.1c00519
[11] Shen, Zhengnan et al. “Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity.” Journal of medicinal chemistry vol. 65,4 (2022): 2940-2955. doi:10.1021/acs.jmedchem.1c01307
[12] Zhao, Yao et al. “High-throughput screening identifies established drugs as SARS-CoV-2 PLpro inhibitors.” Protein & cell vol. 12,11 (2021): 877-888. doi:10.1007/s13238-021-00836-9
[13] Rut, Wioletta et al. “Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design.” Science advances vol. 6,42 eabd4596. 16 Oct. 2020, doi:10.1126/sciadv.abd4596
[14] Delre, Pietro et al. “Repurposing Known Drugs as Covalent and Non-covalent Inhibitors of the SARS-CoV-2 Papain-Like Protease.” Frontiers in chemistry vol. 8 594009. 16 Nov. 2020, doi:10.3389/fchem.2020.594009
[15] Pang, Jie et al. “Discovery of small molecule PLpro inhibitor against COVID-19 using structure-based virtual screening, molecular dynamics simulation, and molecular mechanics/Generalized Born surface area (MM/GBSA) calculation.” Structural chemistry, 1-8. 22 Oct. 2020, doi:10.1007/s11224-020-01665-y
[16] Yuan, Shuofeng et al. “Targeting papain-like protease for broad-spectrum coronavirus inhibition.” Protein & cell, 1–14. 6 Apr. 2022, doi:10.1007/s13238-022-00909-3
[17] Zhang, Linlin et al. “Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors.” Science (New York, N.Y.) vol. 368,6489 (2020): 409-412. doi:10.1126/science.abb3405
[18] Dai, Wenhao et al. “Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease.” Science (New York, N.Y.) vol. 368,6497 (2020): 1331-1335. doi:10.1126/science.abb4489
[19] Jin, Zhenming et al. “Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur.” Nature structural & molecular biology vol. 27,6 (2020): 529-532. doi:10.1038/s41594-020-0440-6
[20] Ma, Chunlong et al. “Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease.” Cell research vol. 30,8 (2020): 678-692. doi:10.1038/s41422-020-0356-z
[21] Li, Jian et al. “Structural Basis of the Main Proteases of Coronavirus Bound to Drug Candidate PF-07321332.” Journal of virology, e0201321. 7 Apr. 2022, doi:10.1128/jvi.02013-21
[22] Owen, Dafydd R et al. “An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19.” Science (New York, N.Y.) vol. 374,6575 (2021): 1586-1593. doi:10.1126/science.abl4784
苏州近岸蛋白质科技股份有限公司,是一家专注于重组蛋白应用解决方案的高新技术企业,主营业务为靶点及细胞因子类蛋白、重组抗体、酶及试剂的研发、生产和销售,并提供相关技术服务。公司定位为医疗健康与生命科学领域原料与技术解决方案的上游供应商,致力于为下游客户提供及时、稳定、优质的产品及服务,助力全球生物医药企业和研究机构的技术与产品创新升级。
详询www.novoprotein.com.cn或致电400-600-0940。